
Synthetic Biology
We are employing engineering principles to model, design and build synthetic gene circuits and programmable cells, in order to create novel classes of diagnostics & therapeutics. We are also using deep learning approaches to discover new genetic parts and enhance the synthetic biology design process.
Antibiotics & AI
As part of the Antibiotics-AI Project, we are harnessing the power of artificial intelligence (AI) to discover novel classes of antibiotics and rapidly understand how they work. We are also using deep learning approaches for the de novo design of new antibiotics and the development of combination treatments.
The Collins Lab is part of the Institute for Medical Engineering and Science (IMES) and the Department of Biological Engineering at MIT, the Harvard-MIT Program in Health Sciences and Technology (HST), the Broad Institute of MIT and Harvard, and the Wyss Institute for Biologically Inspired Engineering at Harvard. At MIT, our lab is part of the Synthetic Biology Center, the Computational and Systems Biology Initiative, and the Microbiology Graduate Program.
RECENT PUBLICATIONS
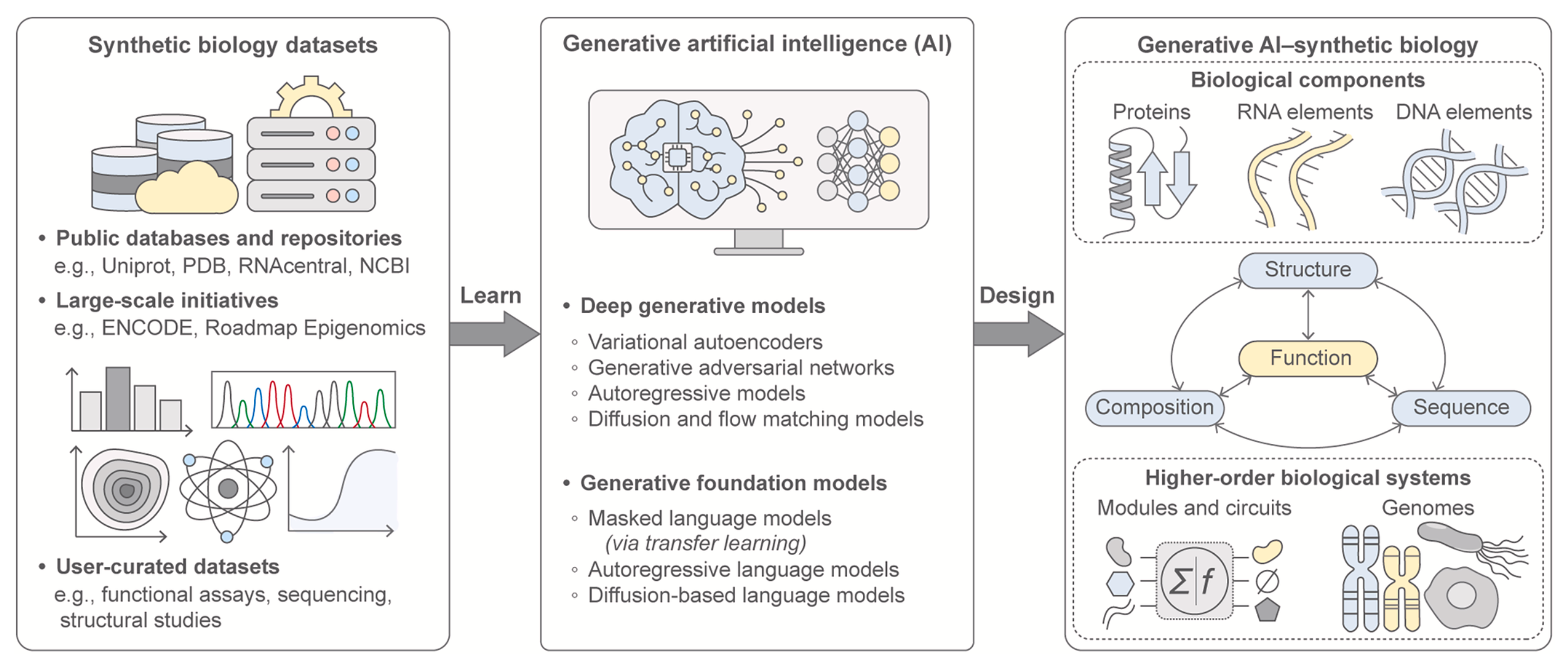
Generative AI for synthetic biology: Designing biological parts, circuits, and genomes
Nayoung Kim, Giuliano De Carluccio, Kehan Zhang, and James J. Collins
Cell Systems (2026)
Synthetic biology aims to achieve predictable, programmable control over living systems by designing and engineering biological components and functions. Over the past 25 years, the field has advanced from foundational molecular tools to increasingly complex systems-level architectures. A new inflection point has emerged with the integration of generative artificial intelligence (AI), catalyzing a fundamental shift in how biological design is conceived and executed. Generative AI now enables the data-driven creation of novel designs with predictable functionality and context-aware precision. Here, we examine the convergence of synthetic biology and generative AI, highlighting key innovations at this emerging frontier of deep generative design across biological parts and systems. We discuss how design frameworks have evolved and outline the opportunities and challenges that lie ahead, spanning biomolecular elements, genetic circuits, and genomes. Finally, we propose a roadmap for how generative AI can unlock a new era of predictable, programmable synthetic biological systems.
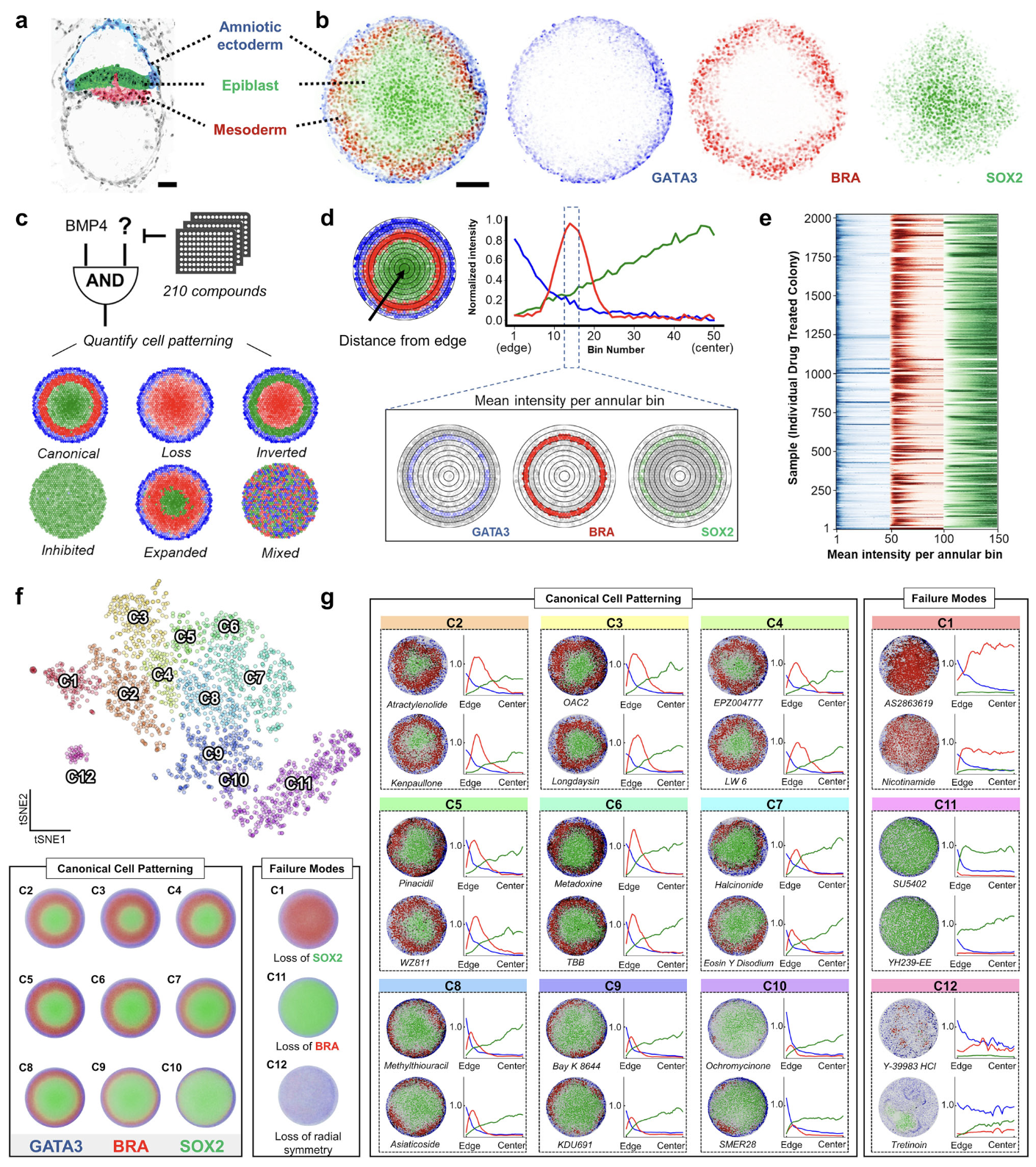
FATE-MAP predicts teratogenicity and human gastrulation failure modes by integrating deep learning and mechanistic modeling
Joseph Rufo, Chongxu Qiu, Dasol Han, Naomi Baxter, Gabrielle Daley, Jasmine Dhillon, Felix Wong, James J. Collins & Maxwell Z. Wilson
Nature Communications (2026)
Gastrulation, a critical developmental stage involving germ layer specification and axes formation, is a major point of failure in human development, contributing to pregnancy loss and congenital malformations. However, due to ethical constraints and anatomical differences in animal models, the failure modes underlying human gastrulation remain poorly understood. To elucidate these failure modes, we introduce FATE-MAP (Failure Analysis and Trajectory Evaluation via Mechanistic-AI Prediction), an integrated platform that combines high-throughput perturbations of human 2D gastruloids with quantitative phenotypic mapping, predictive deep learning, and mechanistic morphogen modeling. Analyzing over 2000 drug-treated human 2D gastruloids, we mapped a phenotypic morphospace that separates canonical patterning, in which primitive-streak fates are correctly specified and radially organized, from failure modes, defined as departures from this organization and marked by a loss of a required fate and/or radial symmetry. To predict and interpret patterning outcomes, FATE-MAP combines a transformer linking chemical structure to phenotype with PDE simulations of morphogen transport and cell fate specification, and projects both outputs onto the experimentally defined morphospace. Applying this framework, we flagged two clinical molecules as potential teratogens and identified two parameters, cell density and SOX2 stability, that form orthogonal morphospace axes along which canonically patterned gastruloids systematically vary. FATE-MAP thus provides a roadmap for decoding human developmental trajectories and accelerating safe therapeutic discovery.
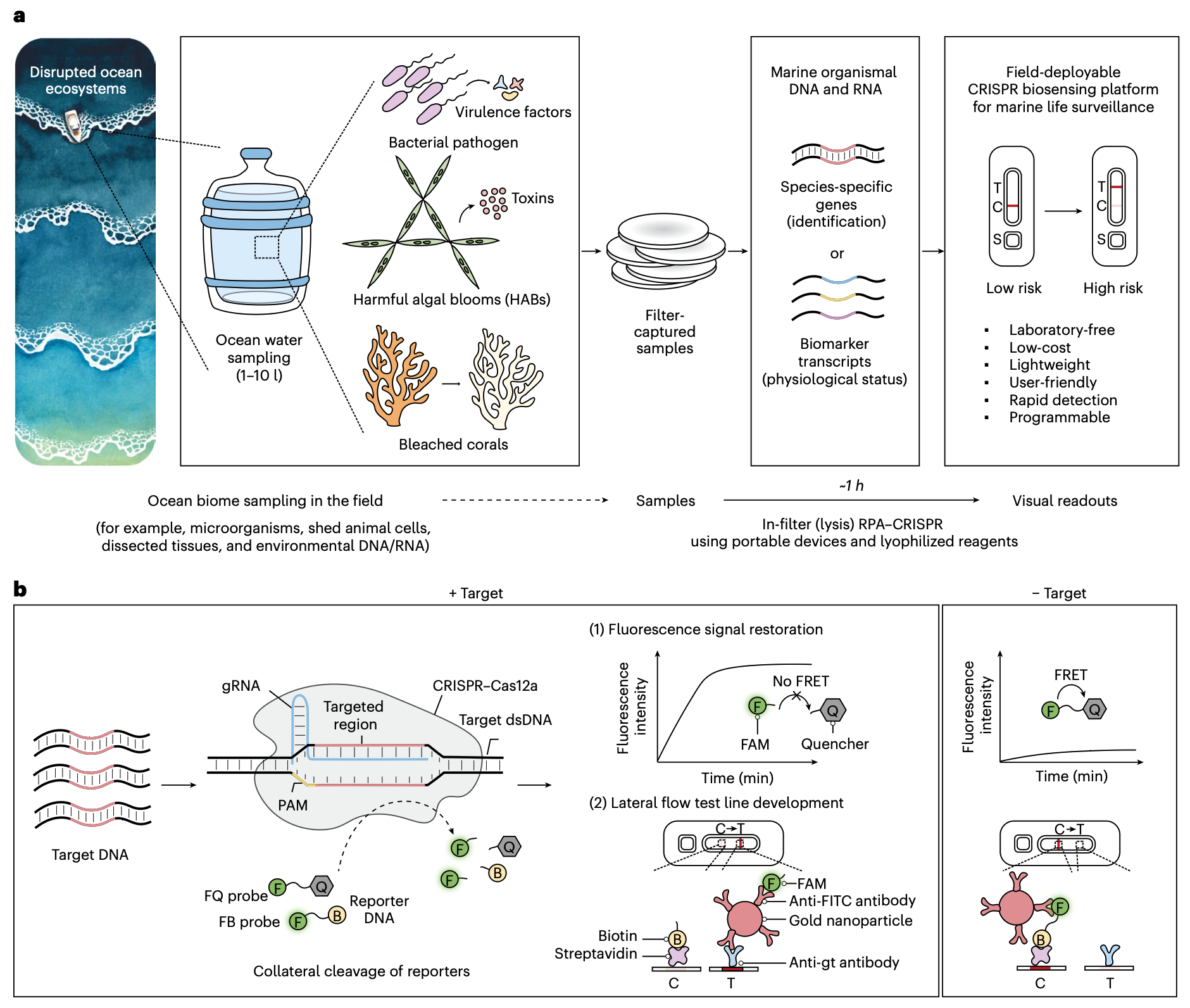
A field‑deployable CRISPR-based biosensing platform for monitoring marine ecosystems
Nayoung Kim, Daniel S. Collins, Nina M. Donghia, Benjamin S. Miller, Hani M. Sallum, Silvi R. Lybbert, Elena Perini, James B. Niemi, James J. Collins and Peter Q. Nguyen
Nature Sustainability (2026)
Ocean ecosystems are undergoing accelerating disruption from human impacts such as climate change. Warming ocean temperatures drive pathogenic outbreaks, increase harmful algal blooms and cause coral stress. These can have serious consequences for marine ecosystems, human health and the aquaculture industry, representing a critical One Health issue. Monitoring key marine species offers valuable insights, but current methods are resource-intensive, low-resolution and unsuitable for frequent deployment. Here we introduce a low-cost, field-deployable CRISPR biosensing platform for detecting marine organismal DNA and RNA. Harnessing the programmability of CRISPR diagnostics for environmental biosurveillance, we demonstrate versatility across three climate-linked indicators: Vibrio spp., Pseudo-nitzschia spp. and heat-stressed corals. Portable 3D-printed processor and incubator devices enable direct processing of filter-captured samples with temperature control. Field readiness is reinforced by lyophilized reagents, lateral flow readouts, dropper-based handling and a two-step multiplexed workflow, delivering results within 1 hour without laboratory instruments. Benchmarking with authentic pathogens and environmental seawater confirmed seawater tolerance and robust detection of 10⁸ colony-forming units per filter of Vibrio pathogens, equivalent to 10² copies per microlitre for 1 litre of filtered sample. This decentralized platform reduces barriers to routine monitoring and can provide early warnings of ecosystem disturbances, while supporting One Health initiatives in the marine space.
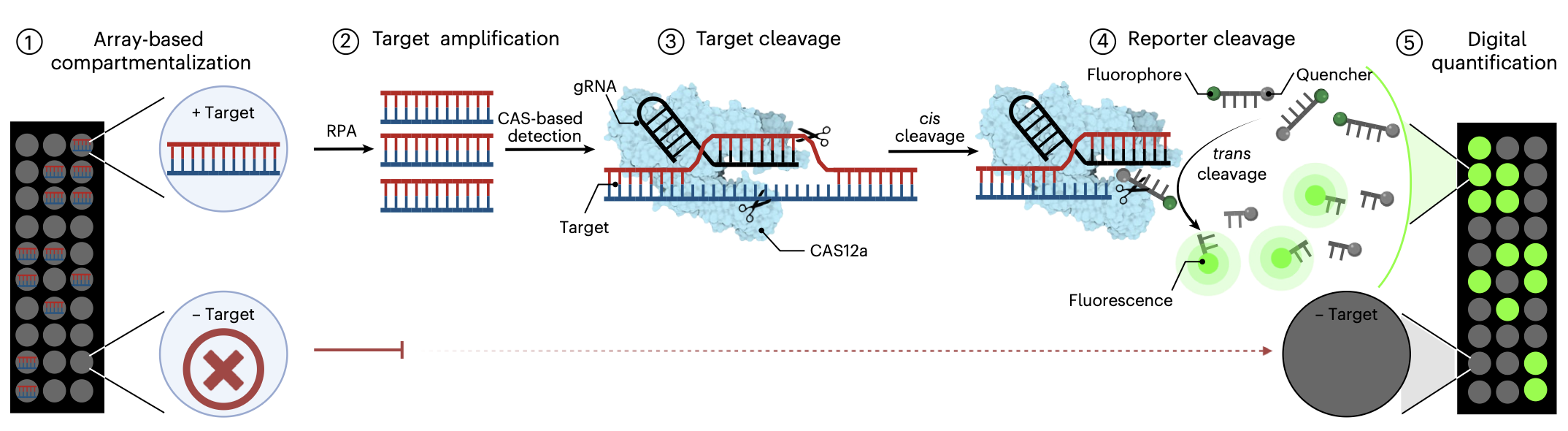
Digital CRISPR-based diagnostics for quantification of Candida auris and resistance mutations
Justin C. Rolando, Anton Thieme, Nicole E. Weckman, Nayoung Kim, Helena de Puig, Xiao Tan, Emily Cotnoir, Vishnu Chaturvedi, James J. Collins and David R. Walt
Nature Biomedical Engineering (2026)
Candida auris, an increasingly prevalent fungal pathogen, requires both rapid identification and antifungal susceptibility testing to enable proper treatment. This study introduces digital SHERLOCK (dSHERLOCK), a platform that combines CRISPR/Cas nucleic acid detection, single-template quantification and real-time kinetics monitoring. Assays implemented on this platform display excellent sensitivity to C. auris from major clades 1–4, while maintaining specificity when challenged with common environmental and pathogenic fungi. dSHERLOCK detects C. auris within 20 min in minimally processed swab samples and achieves sensitive quantification (1 c.f.u. µl⁻¹) within 40 min. To address antifungal susceptibility testing, we develop assays that detect mutations that are commonly associated with azole and echinocandin multidrug resistance. We use machine learning and real-time monitoring of reaction kinetics to achieve highly accurate simultaneous quantification of mutant and wild-type FKS1 SNP alleles in fungal populations with mixed antifungal susceptibility, which would be misdiagnosed as completely susceptible or resistant under standard reaction conditions. Our platform’s use of commercially available materials and common laboratory equipment makes C. auris diagnostics widely deployable in global healthcare settings.

Active learning framework leveraging transcriptomics identifies modulators of disease phenotypes
Benjamin DeMeo, Charlotte Nesbitt, Samuel A. Miller, Daniel B. Burkhardt, Inna Lipchina, Doris Fu, Peter Holderreith, David Kim, Sergey Kolchenko, Artur Szalata, Ishan Gupta, Christine Kerr, Thomas Pfefer, Raziel Rojas-Rodriguez, Sunil Kuppassani, Laurens Kruidenier, Parul B. Doshi, Mahdi Zamanighomi, James J. Collins, Alex K. Shalek, Fabian J. Theis, Mauricio Cortes
Science (2025)
Phenotypic drug screening remains constrained by the vastness of chemical space and technical challenges scaling experimental workflows. To overcome these barriers, computational methods have been developed to prioritize compounds, but they rely on either single-task models lacking generalizability or heuristic-based genomic proxies that resist optimization. We designed an active deep-learning framework that leverages omics to enable scalable, optimizable identification of compounds that induce complex phenotypes. Our generalizable algorithm outperformed state-of-the-art models on classical recall, translating to a 13-17x increase in phenotypic hit-rate across two hematological discovery campaigns. Combining this algorithm with a lab-in-the-loop signature refinement step, we achieved an additional two-fold increase in hit-rate and molecular insights. In sum, our framework enables efficient phenotypic hit identification campaigns, with broad potential to accelerate drug discovery.