
Synthetic Biology
We are employing engineering principles to model, design and build synthetic gene circuits and programmable cells, in order to create novel classes of diagnostics & therapeutics. We are also using deep learning approaches to discover new genetic parts and enhance the synthetic biology design process.
Antibiotics & AI
As part of the Antibiotics-AI Project, we are harnessing the power of artificial intelligence (AI) to discover novel classes of antibiotics and rapidly understand how they work. We are also using deep learning approaches for the de novo design of new antibiotics and the development of combination treatments.
The Collins Lab is part of the Institute for Medical Engineering and Science (IMES) and the Department of Biological Engineering at MIT, the Harvard-MIT Program in Health Sciences and Technology (HST), the Broad Institute of MIT and Harvard, and the Wyss Institute for Biologically Inspired Engineering at Harvard. At MIT, our lab is part of the Synthetic Biology Center, the Computational and Systems Biology Initiative, and the Microbiology Graduate Program.
RECENT PUBLICATIONS
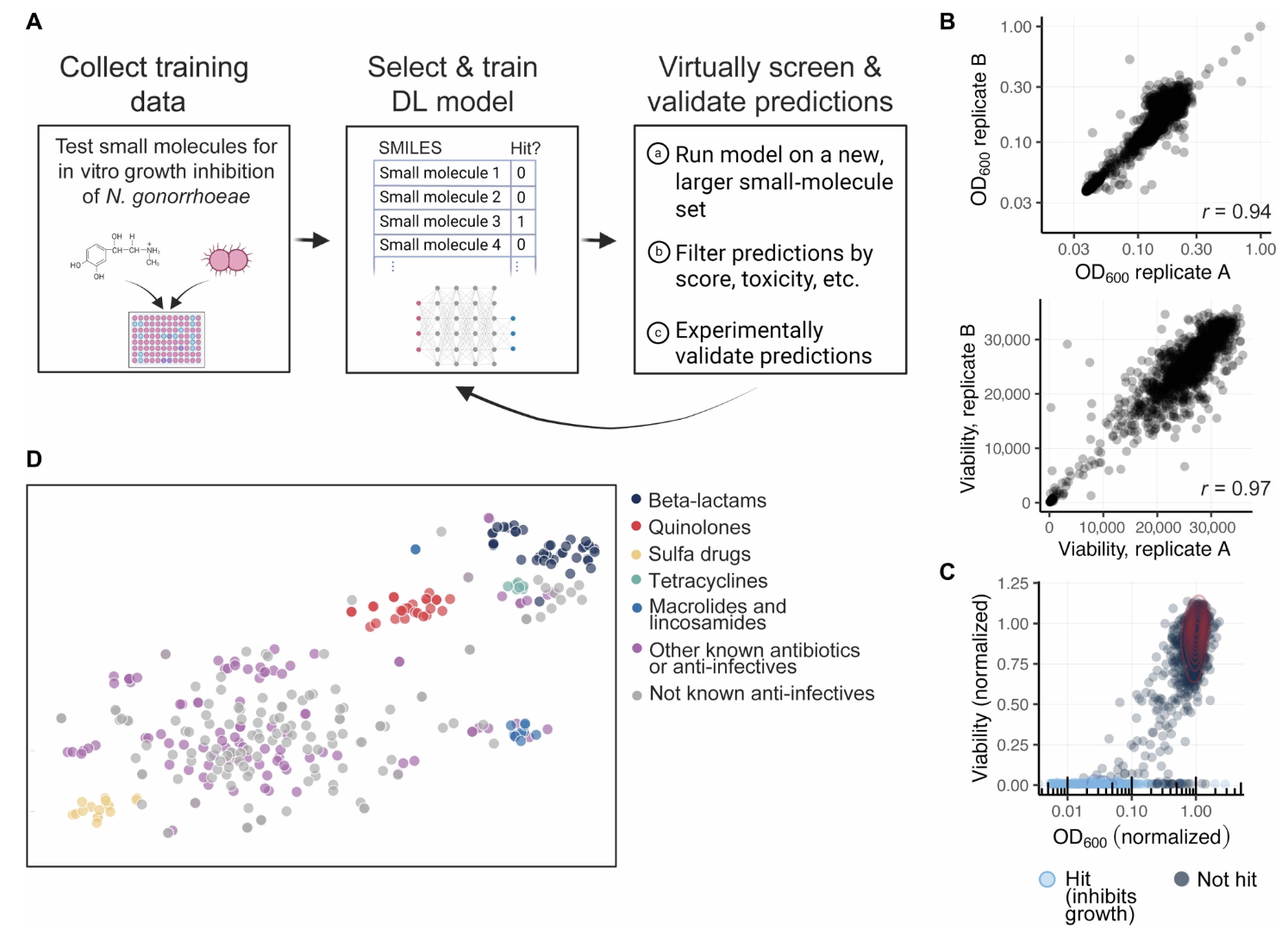
Deep learning–enabled discovery of antibiotics effective against Neisseria gonorrhoeae
Melis N. Anahtar, Jacqueline A. Valeri, Seyed Majed Modaresi, Aarti Krishnan, Nina M. Donghia, Samantha G. Palace, Erica J. Zheng, Aakanksha Gulati, Alicia Jorgenson, Abidemi Junaid, Parijat Bandyopadhyay, Andreas Luttens, Krishna Suresh, Paige Edwards, Felix Wong, Yu Zhang, Danilo Ritz, Margaux Gaborieau, Edmund Loh, Massimiliano Gaetani, Marie-Stephanie Aschtgen, Amir Ata Saei, Yonatan H. Grad, Donald E. Ingber, James J. Collins.
Science Translational Medicine (2026)
Neisseria gonorrhoeae is a common Gram-negative pathogen with increasing resistance to all recommended antibiotics. There is a critical need to improve the efficiency of the antibiotic hit discovery process to replenish the drug development pipeline. Here, we show that deep learning models can augment high-throughput screens to identify readily available molecules with narrow-spectrum activity against difficult-to-treat strains of N. gonorrhoeae. We phenotypically tested 38,650 small molecules for N. gonorrhoeae growth inhibition to train a predictive graph neural network (GNN) model. We benchmarked the model’s performance against other architectures, including a large language model, and found that GNNs more accurately identify active, drug-like molecules that are structurally distinct from the training set and known antibiotics. Using the model to virtually screen ~6 million compounds, we identified 213 compounds for experimental validation and found that 83 (39%) inhibited N. gonorrhoeae growth. Two of these compounds were structurally dissimilar to existing antibiotics, maintained potency against multidrug-resistant N. gonorrhoeae strains in vitro, exhibited promising selectivity indices, and were rapidly bactericidal with low frequencies of resistance. Proteomic studies revealed their distinct mechanisms of action, with one compound targeting alanine racemase, an enzyme involved in the essential process of peptidoglycan synthesis. Furthermore, the compounds showed early promise in reducing N. gonorrhoeae titers in a human vagina-on-a-chip infection model and a mouse vaginal infection model. Our work establishes the deep learning–enabled discovery of selective antibacterial compounds against N. gonorrhoeae as a much-needed hit discovery tool to address the growing crisis of antimicrobial resistance for this pathogen.
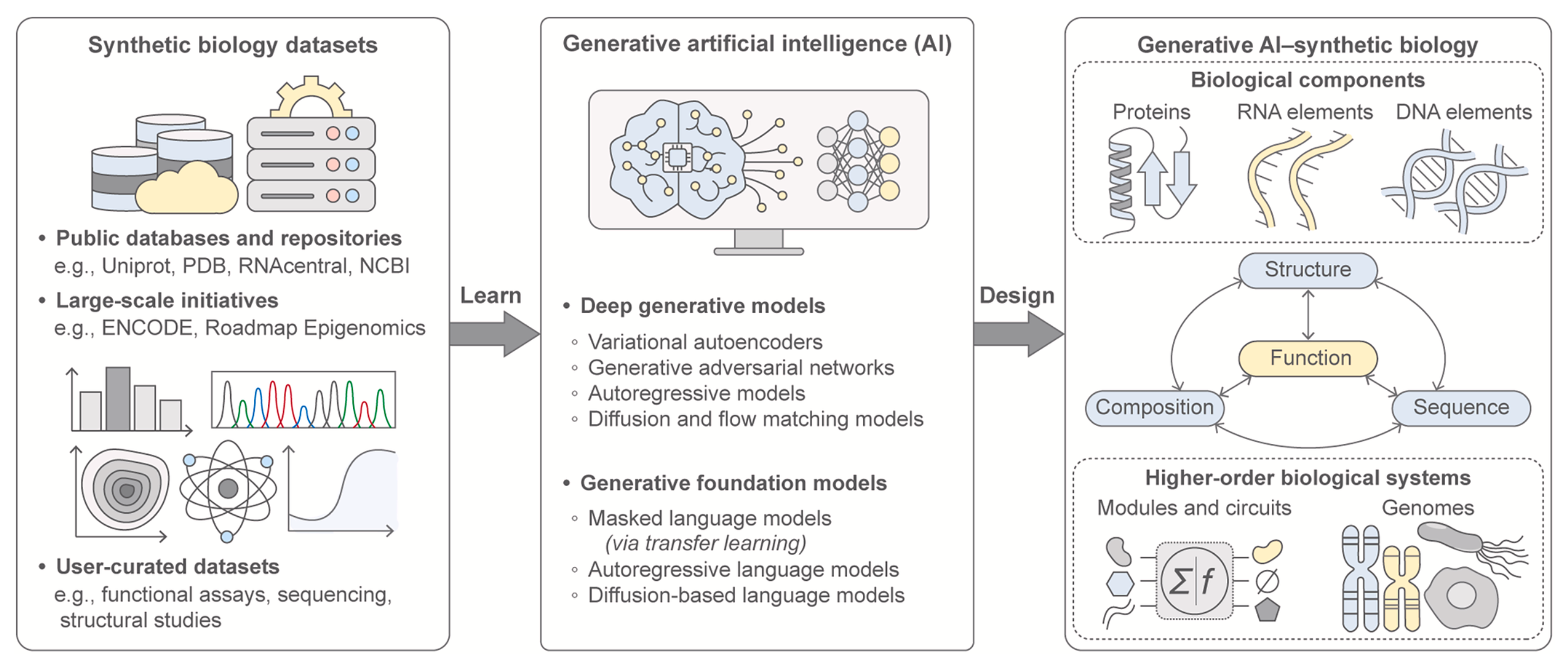
Generative AI for synthetic biology: Designing biological parts, circuits, and genomes
Nayoung Kim, Giuliano De Carluccio, Kehan Zhang, and James J. Collins
Cell Systems (2026)
Synthetic biology aims to achieve predictable, programmable control over living systems by designing and engineering biological components and functions. Over the past 25 years, the field has advanced from foundational molecular tools to increasingly complex systems-level architectures. A new inflection point has emerged with the integration of generative artificial intelligence (AI), catalyzing a fundamental shift in how biological design is conceived and executed. Generative AI now enables the data-driven creation of novel designs with predictable functionality and context-aware precision. Here, we examine the convergence of synthetic biology and generative AI, highlighting key innovations at this emerging frontier of deep generative design across biological parts and systems. We discuss how design frameworks have evolved and outline the opportunities and challenges that lie ahead, spanning biomolecular elements, genetic circuits, and genomes. Finally, we propose a roadmap for how generative AI can unlock a new era of predictable, programmable synthetic biological systems.
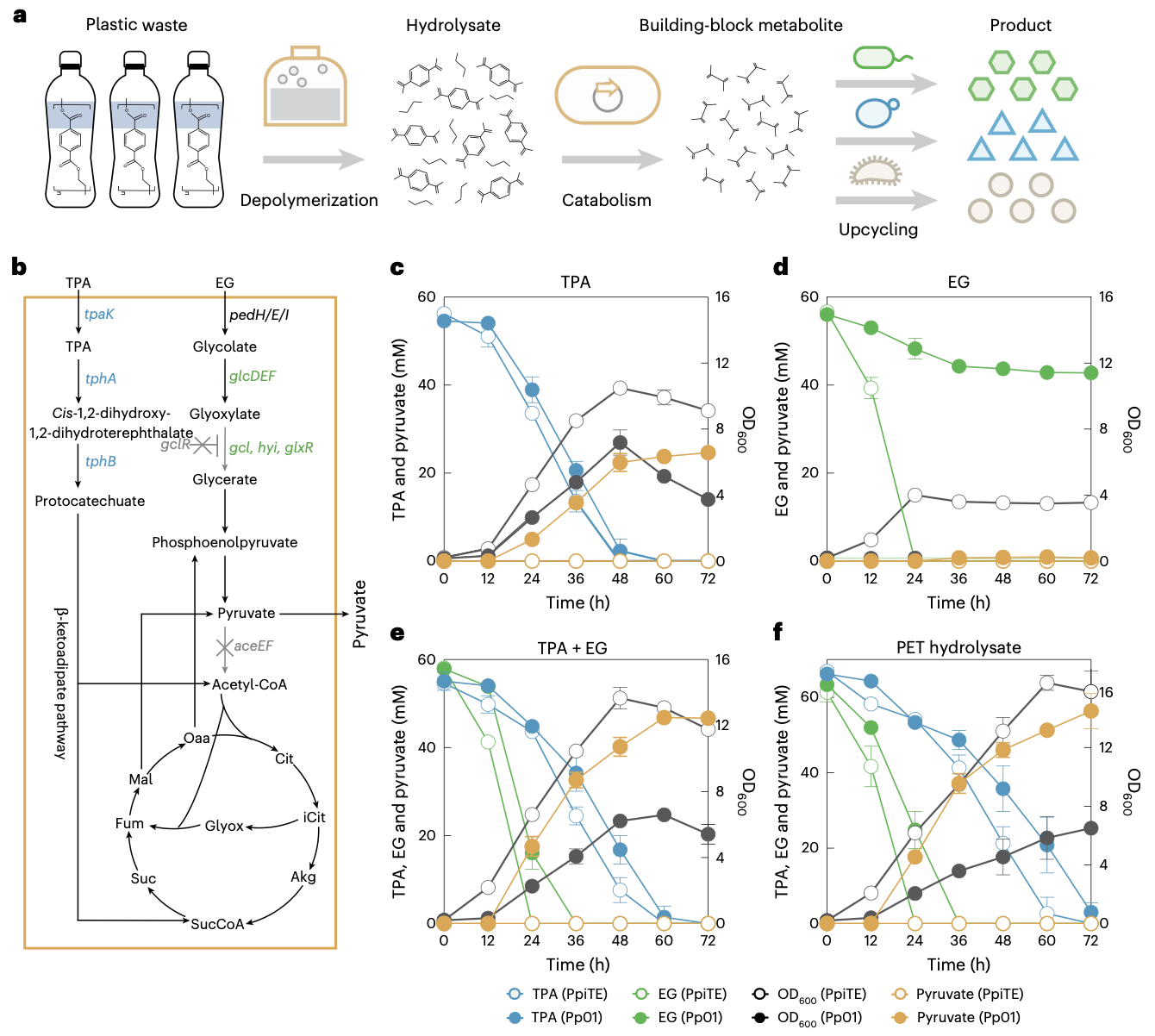
A programmable microbial assembly line for plastic upcycling
Yuanchao Qian, James J. Collins and Ting Lu
Nature Sustainability (2026)
Biological upcycling is a promising strategy for handling end-of-life plastics. However, current practices, which predominantly focus on yielding fixed single outputs, face challenges in meeting diverse and evolving product demands. Here we present a programmable microbial assembly line that enables the efficient and versatile upcycling of plastic waste into various end products. The assembly line involves a deconstruction strain that catabolizes pretreated polyethylene terephthalate into pyruvate, serving as a universal building block, and a set of interchangeable production strains that transform this building block into tailorable outputs. Such a design allows a rapid, combinatorial assembly of microbial strains into diverse biomanufacturing systems. Accordingly, programmable upcycling is achieved, yielding successful production of a wide range of final products, including chemicals, fuels, enzymes, biopolymers and electricity. This work establishes a reconfigurable valorization platform, providing insights into biological treatment of plastic waste as well as into need-driven, sustainable biomanufacturing.
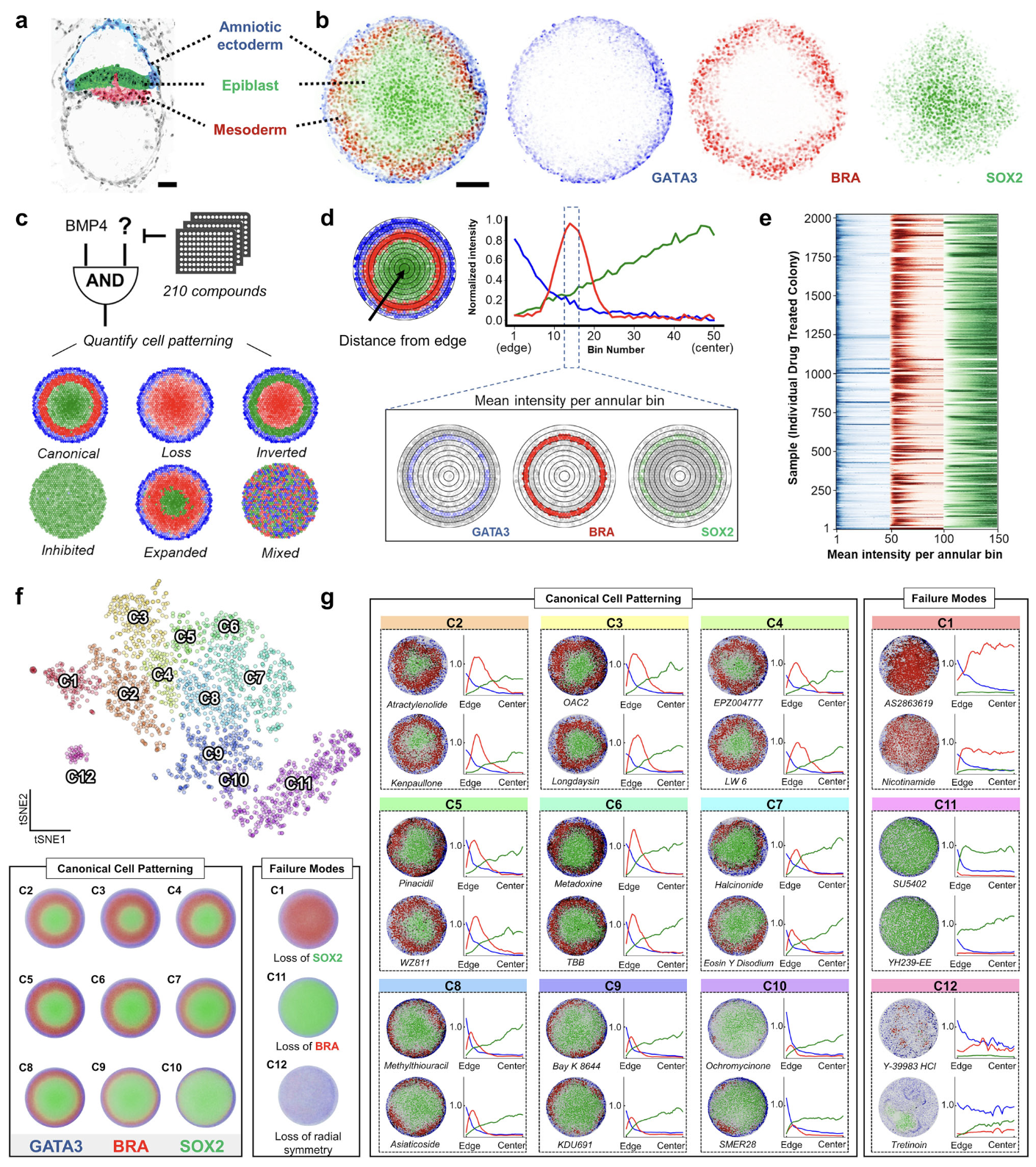
FATE-MAP predicts teratogenicity and human gastrulation failure modes by integrating deep learning and mechanistic modeling
Joseph Rufo, Chongxu Qiu, Dasol Han, Naomi Baxter, Gabrielle Daley, Jasmine Dhillon, Felix Wong, James J. Collins & Maxwell Z. Wilson
Nature Communications (2026)
Gastrulation, a critical developmental stage involving germ layer specification and axes formation, is a major point of failure in human development, contributing to pregnancy loss and congenital malformations. However, due to ethical constraints and anatomical differences in animal models, the failure modes underlying human gastrulation remain poorly understood. To elucidate these failure modes, we introduce FATE-MAP (Failure Analysis and Trajectory Evaluation via Mechanistic-AI Prediction), an integrated platform that combines high-throughput perturbations of human 2D gastruloids with quantitative phenotypic mapping, predictive deep learning, and mechanistic morphogen modeling. Analyzing over 2000 drug-treated human 2D gastruloids, we mapped a phenotypic morphospace that separates canonical patterning, in which primitive-streak fates are correctly specified and radially organized, from failure modes, defined as departures from this organization and marked by a loss of a required fate and/or radial symmetry. To predict and interpret patterning outcomes, FATE-MAP combines a transformer linking chemical structure to phenotype with PDE simulations of morphogen transport and cell fate specification, and projects both outputs onto the experimentally defined morphospace. Applying this framework, we flagged two clinical molecules as potential teratogens and identified two parameters, cell density and SOX2 stability, that form orthogonal morphospace axes along which canonically patterned gastruloids systematically vary. FATE-MAP thus provides a roadmap for decoding human developmental trajectories and accelerating safe therapeutic discovery.
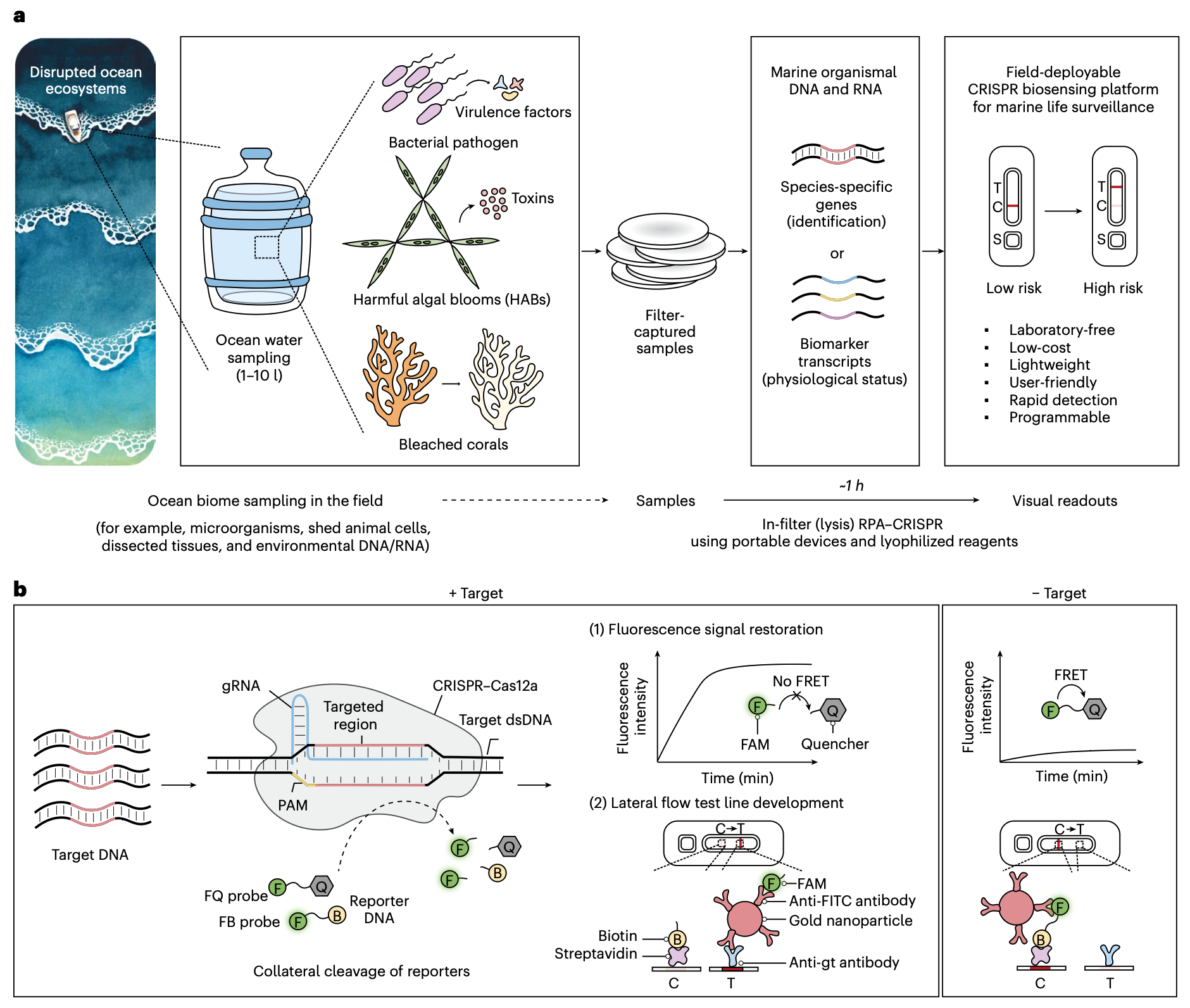
A field‑deployable CRISPR-based biosensing platform for monitoring marine ecosystems
Nayoung Kim, Daniel S. Collins, Nina M. Donghia, Benjamin S. Miller, Hani M. Sallum, Silvi R. Lybbert, Elena Perini, James B. Niemi, James J. Collins and Peter Q. Nguyen
Nature Sustainability (2026)
Ocean ecosystems are undergoing accelerating disruption from human impacts such as climate change. Warming ocean temperatures drive pathogenic outbreaks, increase harmful algal blooms and cause coral stress. These can have serious consequences for marine ecosystems, human health and the aquaculture industry, representing a critical One Health issue. Monitoring key marine species offers valuable insights, but current methods are resource-intensive, low-resolution and unsuitable for frequent deployment. Here we introduce a low-cost, field-deployable CRISPR biosensing platform for detecting marine organismal DNA and RNA. Harnessing the programmability of CRISPR diagnostics for environmental biosurveillance, we demonstrate versatility across three climate-linked indicators: Vibrio spp., Pseudo-nitzschia spp. and heat-stressed corals. Portable 3D-printed processor and incubator devices enable direct processing of filter-captured samples with temperature control. Field readiness is reinforced by lyophilized reagents, lateral flow readouts, dropper-based handling and a two-step multiplexed workflow, delivering results within 1 hour without laboratory instruments. Benchmarking with authentic pathogens and environmental seawater confirmed seawater tolerance and robust detection of 10⁸ colony-forming units per filter of Vibrio pathogens, equivalent to 10² copies per microlitre for 1 litre of filtered sample. This decentralized platform reduces barriers to routine monitoring and can provide early warnings of ecosystem disturbances, while supporting One Health initiatives in the marine space.